The synthetic origin theory of SARS-CoV-2
Why it still stands, and what it would take to disprove it.

The pre-print by Drs. Valentin Bruttel, Tony VanDongen and myself found strong evidence consistent with a synthetic origin of SARS-COV-2, generating considerable scientific discussion.
There are critiques, but for every critique there is a solid defense and so our theory still stands. The two main critiques are:
Other methods to assemble a virus exist and some people like those other methods
Recombination could explain this pattern
Big picture:
Other methods for in vitro genome assembly exist, but they weren’t commonly used to make coronavirus infectious clones prior to COVID-19. The method we study was used in 80% of the infectious clones we found in our meta-analysis.
Recombination can explain anything, so as a competing hypothesis it needs to be clarified. After all, recombination could explain a fluorescent green rat, but that doesn’t mean natural recombination is the most likely explanation, especally if the rat is found near a research center known to insert GFPs into animals. Researchers proposing “recombination” need to do more to flesh out the odds of their hypothesis.
Below is the more detailed reason why the critiques have failed to disprove our theory.
Other methods of assembly exist
To say “other methods of assembly exist, so therefore the method proposed was not used for SARS-CoV-2” is like walking up to a murder scene and saying “there are more efficient ways to murder someone, I wouldn’t have chosen this method, so therefore it wasn’t a murder.” At the heart of this critique is an assumption that the method for in vitro genome assembly we study was unlikely or uncommon.
To critically examine this claim, we need to understand whether the particular method of viral assembly we studied was common. Our study conducted a meta-analysis to find all CoV infectious clones from 2000-2019. We searched for all studies containing the terms “coronavirus”, “infectious clone”, and “type IIS”. The proposed other-methods-of-assembly (Golden Gate assembly) also utilizes type IIS restriction enzymes, so such infectious clones would have been found in our analysis.
Our meta-analysis yielded 10 examples of infectious clones:
Of the 10 examples above, only PEDV and MHV were assembled using golden gate assembly. The other 8 out of 10 CoV infectious clones we found from 2000-2019 were assembled by the method we’ve described. Type II directional cloning was by far the most common method for assembling coronavirus genomes in vitro prior to COVID-19.
Notably, one of these CoVs was even assembled in Wuhan: WIV1 was assembled in the Wuhan Institute of Virology with PI’s Shi Zheng-Li and Peter Daszak. rWIV1 was assembled adding/removing BglI sites with silent mutations (black/white arrows below, respectively), utilizing some pre-existing sites (no arrows), and then proceeding with exactly the method of type II directional cloning we examine in our paper.
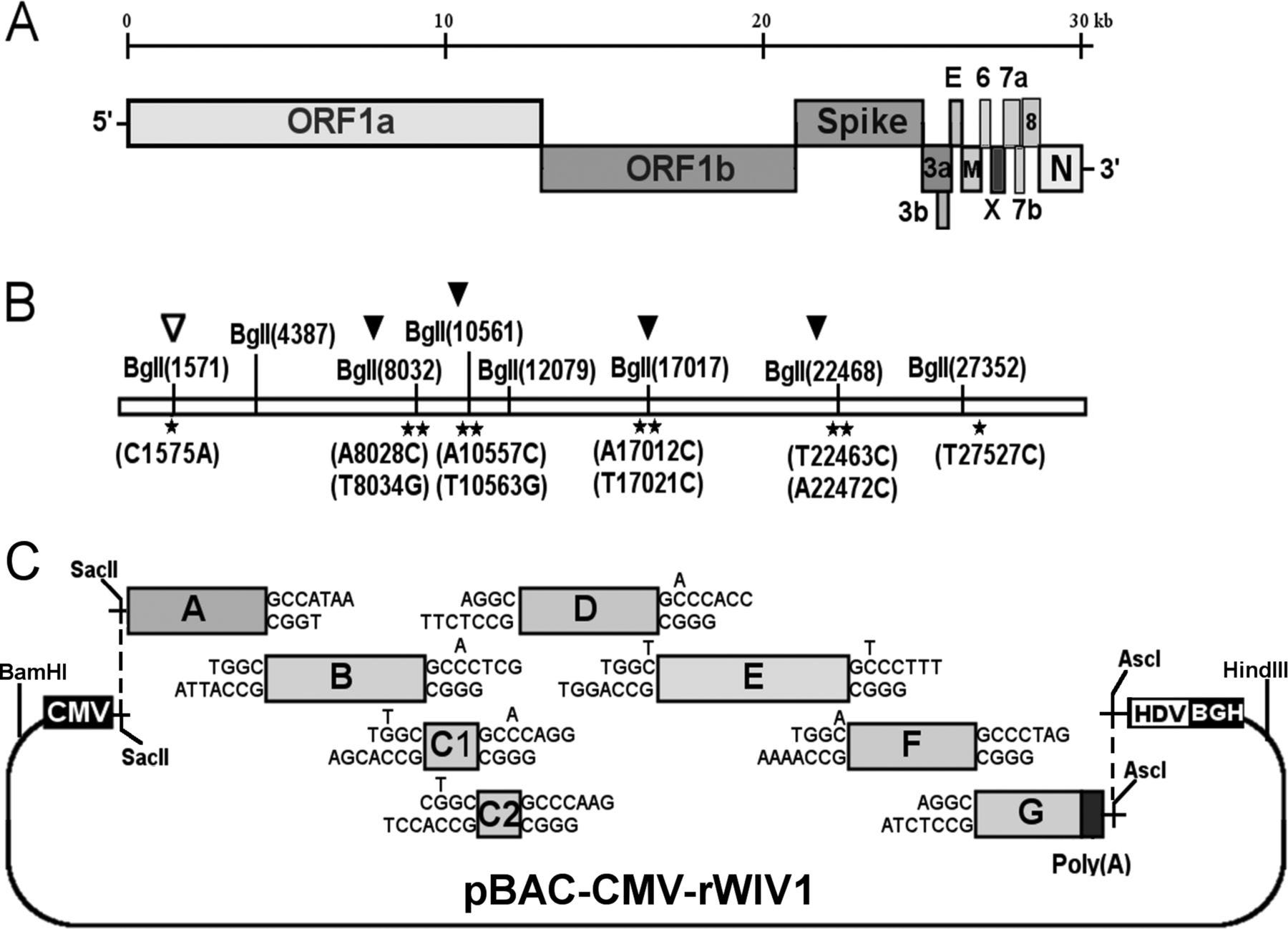
Figure 2 of our paper includes WIV1, above, as well as the MERS-CoV, as an illustration of the fingerprint one would, in fact, find on the infectious clones.

While other methods exist, type II directional cloning was used in 80% of the infectious clones we found in our meta-analysis and it was specifically used in the Wuhan Institute of Virology in a collaboration with EcoHealth alliance to study chimeric bat coronaviruses.
To better understand the design & variety of infectious clones built by type II directional assembly, it’s worth looking closer at how these clones were used.
After making their infectious clones, researchers would often add glowing proteins to specific parts of the virus to study when, where, and how various components of the virus are produced. Another very common experiment was to swap-parts of viruses, like putting GI Joe arms on a Mr. Potato Head doll to see if GI Joe Arms help Mr. Potato Head gain the function of lifting weights. Researchers used the MERS-CoV infectious clone above to modify the Spike protein & measure receptor-binding. The study modifying both SARS-CoV Urbani and the bat CoV WIV1 put the WIV1 Spike gene inside a SARS-CoV Urbani backbone, and doing so required restriction sites that were shared by both SARS-CoV and WIV1.
Swapping viral parts was a common research project. When swapping parts of viruses in the lab with reveres genetic systems, it’s much easier to swap parts if there are pre-existing, conserved restriction sites. If Mr. Potato Head had cutting sites at the shoulders but GI Joe only had cutting points on his wrist, researchers would add cutting points at the shoulders of GI Joe to swap parts. Researchers would add the appropriate restriction sites corresponding to restriction sites found in another virus, and so conserved restriction sites were valuable features for making chimeric viruses. The existence of the same BsaI/BsMBI restriction sites in SARS-CoV-2 and in other CoVs has been proposed as evidence that these sites are of natural origin, but a backbone containing restriction sites also found in other CoVs is also consistent with a laboratory origin as it enables the assembly of chimeric CoVs.
Recombination could explain this
Genetic recombination is the swapping of genetic parts. Recombination could explain the WIV1 spike protein inserted into the SARS-CoV Urbani backbone, but what is the likelihood of that in nature? All of genetic engineering involves mutation and recombination, the same processes at play in evolution, so “recombination” is too general as it can explain anything, even things that were clearly made in a lab. Observing that recombination occurs in nature does not refute our hypothesis, it just introduces another hypothesis that needs to be further studied & better quantified.
Imagine you were walking around in Bergen, Norway and came across the green fluorescent mice below. You examine their genomes and find a jellyfish gene encoding the glowing protein. Technically, that could be explained by recombination, but is that likely? After all, there’s a lab at the University of Bergen just a mile or so away that has inserted jellyfish genes into mice before. SARS-CoV-2 emerges in Wuhan, a global hotspot of coronavirus research, and it has a type II restriction map that is an idealized reverse genetic system unlike anything found before in a wild CoV. That could be explained by recombination, but is that likely?

The recombination hypothesis is a hypothesis. We didn’t observe a recombination event. Rather, researchers at the Chinese National Academy, and Eddie Holmes, reportedly found sequences from bats in a botanical garden in Yunnan China. They combine their 82 new sequencing libraries from the botanical garden bats with 18 additional sequences described earlier & from the same group. An important sidenote is that Eddie Holmes is under investigation by the US Congress Oversight & Reform Committee examining apparent conflicts of interest, suppression of scientific discourse, and other matters of importance - the results of that investigation may impact the trustworthiness of sequences behind the hypothesized recombination.
Anyways, many new sequences uploaded from one group. The new sequences contain two curiosities - RpYN06 and RmYN02 which appear to be more similar to SARS-CoV-2 in some places of the genome than others. This pattern of similarity lends itself to the hypothesis of a recombination event, contingent on the veracity of the sequences and with confidence related to how much additional data we’ve collected to corroborate this hypothesis.

Recombination is common in RNA viruses. Recombination is how H1N1 can turn into H1N2, it’s why Ebola has a funky “VP35” gene also found in bats, and more. We rarely observe recombination in real-time, but rather we analyze sequences with particular algorithms and those algorithms output hypothesized recombination sites. The figure above by Temmam et al. shows the output of a particular method for analyzing genomes and classifying regions as recombinant - regions classified as recombinant (by this method) are labeled by the most closely related sequence for each hypothesized recombinant region.
The exact method is worth examining closely. Temmam et al utilized a 2006 software package called IDPlot. The researchers grabbed 106 genomes, whittled them down to 36 genomes of interest, and then input these 36 genomes into IDPlot. Temmam et al. don’t describe the input arguments used for IDPlot, and those matter as they may determine whether these estimated recombination sites are conservative or if there’s likely a high false-positive rate and low confidence in any one site. The estimated breakpoints for recombination are just estimates. Temmam et al. don’t provide the uncertainty of their estimates, either on breakpoints or odds that the recombination event is inferred by chance, so we have no way of knowing if the algorithm was highly confident or highly uncertain about the hypothesized recombination events.
The recombination hypothesis relies on these hypothesized recombination events, and these hypothesized events are of unknown certainty, derived from sequences collected by one lab. The recombination hypothesis says that the ancestral sequence of SARS-CoV-2 probably looked like RpYN06 in this 5th fragment from 7,117 to 11,462. Tony VanDongen made the restriction maps for comparison below. In the region of interest from 7,117-11,462, RpYN06 has the BsmBI restriction site found in SARS-CoV-2 and lacks the two conserved BsaI sites. The hypothesized recombination event could provide an alternative explanation for the SARS-CoV-2 restriction map at this narrow range, but not the rest of SARS-CoV-2. Furthermore, the recombination hypothesis is a hypothesis whose probabilities we don’t know because Temmam et al. haven’t included the uncertainty estimates in their paper.
In this hypothesized region of recombination, there are 77 mutations in 4,345 nucleotides, a rate of mutation per nucleotide that would be equal to 530 mutations over the whole genome. BANAL-52, the closest relative to SARS-CoV-2 at the whole-genome level, differs by SARS-CoV-2 by 903 mutations. SARS-CoV-2 evolves at about 2 mutations per month, so 900 mutations separating two branches, with ~451 mutations on each branch, would imply approximately 19 years of evolution. The RpYN06 recombination hypothesis, meanwhile, implies 11 years of evolution since the recombination event. This is a testable hypothesis: if researchers produce a coronavirus in a bat that branched off from SARS-CoV-2 by under 11 years, then under the RpYN06 recombination hypothesis that sequence ought to look like RpYN06 and not BANAL52 in this region.
Recombination can explain a small part of the SARS-CoV-2 restriction map, but there are alternative explanations too. A close examination of the sequencing libraries producing RpYN06 and RmYN02 found significant evidence of contamination. The sequences themselves may not be correct - such uncertainty needs to be taken int account when evaluating the probabilities of the recombination hypothesis.
Of note, Pekar et al. claimed to have found evidence of multiple spillovers. Eddie Holmes, Alex Crits-Cristoph and others advancing the recombination hypotheses are co-authors on that paper. Their paper claims to have estimated two big lineages at the base of the SARS-CoV-2 evolutionary tree, but they discarded intermediate sequences that contradicted this finding. In their words, “We identified numerous instances of C/C and T/T genomes sharing rare mutations with lineage A or lineage B viruses, often sequenced in the same laboratory, indicating that these intermediate genomes are likely artifacts of contamination or bioinformatics”. So, the same researchers who claim that RpYN06 and RmYN02 must be included as definitive evidence of recombination - despite these sequences coming from the same lab and strong evidence of contamination - had in their own work removed sequences because they came from the same lab and believed there was evidence of contamination.
There seem to be different standards applied depending on whether a study supports or contradicts the authors’ favored theory.
While we are highly uncertain about lab artifacts, we nonetheless generously examined the RpYN06 recombination hypothesis by assuming the RpYN06 recombination explains the three restriction sites in that region of recombination. Doing so doesn’t change our main results. The SARS-CoV-2 BsaI/BsmBI map is still anomalous among natural CoVs and consistent with known infectious clones assembled by the most common pre-COVID methods of in vitro assembly. All BsaI/BsmBI restriction sites still deviate from close relatives by exclusively silent mutations, and there is still a higher concentration of silent mutations within BsaI/BsmBI sites than the rest of the genome (OR=7.1 P=1.6e-5 for RaTG13, OR=3 P=0.09 for BANAL52). While P=0.09 is only marginally significant for BANAL52, the odds-ratio for BANAL52 is in the same direction of RaTG13 and the P-value for RaTG13 is extremely significant. It would be cherry-picking to rest entirely on one or the other, and furthermore these P-values are not necessarily the correct ones but rather are the conditioned on the hypothesis that SARS-CoV-2 had exactly the same restriction map of RpYN06 in the hypothesized region of recombination.
The Synthetic Origin Theory Stands
Our theory that SARS-CoV-2 may have been synthesized by type II directional assembly stands. Recombination is an alternative hypothesis whose odds have not been fully fleshed out by those proposing it, and these hypotheses can be tested.
If an independent group without conflicts of interest on the lab origin question found a virus with less than 11 years of divergence from SARS-CoV-2, its genome can be examined for the hypothesized recombination event as well as an updated assessment of mutations at BsaI/BsmBI sites. Researchers can assess if RaTG13, RpYN06, and RmYN02 are, in fact, actual viruses and not simply letters on a screen whose weird features are due to contamination, lab-specific artifacts, or other technical issues. Independent researchers may find a relative of RpYN06 that could corroborate (or reject) the lineage proposed to have caused so many recombination events in so little time. Additional sequences around RpYN06 can also give change our certainty about the recombination hypothesis and whether a proposed recombination from this lineage had a BsaI/BsmBI map like RpYN06 or not. Perhaps RpYN06 is an anomaly in its lineage, and other viruses closely related to RpYN06 contain BsaI/BsmBI sites identical to BANAL52 or RaTG13 in this region.
Our main finding remains uncontested: the SARS-CoV-2 BsaI/BsmBI sites are an anomaly in nature and consistent with a reverse genetic system. Recombination might change the evolutionary story slightly, but it doesn’t reject our theory. Since recombination can explain anything, testing these competing hypotheses requires more data and future research. We have fleshed out our theory, provided the likelihoods of meeting all the criteria for reverse genetic systems as well or better than SARS-CoV-2. It is still a valid theory that SARS-CoV-2 may have been assembled in vitro by common pre-COVID methods used in Wuhan.
We thank researchers for their engagement on this issue, and encourage future work to clarify the recombination hypothesis, including quantifying their uncertainty about the recombination event, examining the restriction sites of other viruses proposed to have recombined with SARS-CoV-2, and ensuring the sequences in question are not invalidated by peer-reviewed reports of contamination in those sequences.
Science goes on.
Excellent, Alex! Keep up the great work!
Not only did SARS-2 emerge right next to the WIV (nothing to see here), it showed up highly adept to human-human transmission, as if it took a mysterious pit-stop before making its debut...
Love your work, Alex, and the issue-focused (not person-focused), gracious way you articulate your position. Thank you!