The case for a lab origin of SARS-CoV-2
I used to believe it was spillover, but now I believe SARS-CoV-2 came from a lab

The Global Jury
Ladies and gentlemen of our global jury, thank you for taking the time out of your lives to be here today and evaluate this case on the origins of a pandemic virus.
I know you all are busy living fulfilling lives, advancing your careers, and building your skills and knowledge far from this previously esoteric field of virology. I know you all were doing your jobs prior to COVID, you all suffered through the pandemic together, some of you lost loved ones. Now, here we are. I know it’s not easy to learn new tricks from somebody else’s job, and I know this case is uniquely challenging because understanding the evidence and honoring our civic duty requires we all take crash courses in biology, virology, data science, and other fields of science, each of which is required to understand and contextualize the evidence before us. As surely as ultraviolet light is invisible to our naked eyes but its existence and cause of cancer evident to modern science, the evidence of a research-related origin of SARS-CoV-2 exists, difficult to see by the untrained mind yet, with training, the evidence indisputably reveals a research-related origin of SARS-CoV-2 beyond reasonable doubt.
The SARS-CoV-2 pandemic has disrupted all of our lives. Over 18 million people worldwide are confirmed dead. Over 60 million extra people faced acute hunger. Over 100 million children were thrust into multidimensional poverty, and more. The fallout will persist for years. The inequitable declines in educational attainment may have life-long consequences for our kids, and the steady increases in excess mortality from disruptions in routine care and disruptions to our daily lives may continue to take loved ones from us or the rest of our lives. The case before us today is historic. We can only hope this be the most important case in our lives, and that no other case involves so much injury and such clear evidence of scientists taking risks that were unacceptable to society at large. As we stand amidst the wreckage today, we have a duty to future generations to help them understand what caused a pandemic. Your service in our global jury, your acquisition of new knowledge to help us understand this critical evidence, will help us create a better world in which catastrophic pandemics are less-likely, in which science continues to advance our civilization with accountability for risky research that goes wrong. Accountability is necessary to regulate risky research and prevent the reckless endangerment of our civilization.
The first step towards accountability is realizing that SARS-CoV-2 came from a lab.
While I’m deeply saddened by the inexplicable magnitude of human suffering caused by this pandemic, I’m serenely honored to be inextricably bound to this moment in history, and to be examining this issue with such a brilliant global jury brought together by the internet, itself a product of science. This is our moment in history. You all have shown a remarkable aptitude as quick studies of novel concepts, from the genetic code and modern methods of bioengineering to evolutionary trees, cell-phone datasets, and more. If I were a dean of Harvard, I would be giving you all honorary degrees as experts on the topic of SARS-CoV-2 origins. Thank you, profoundly, for fulfilling your civic duty & helping us learn the lessons from this historic accident.
Let’s cut to the case.
The Zoonotic Theory
The only alternative to a lab origin for SARS-CoV-2 is the “zoonotic” theory that the virus existed in wildlife populations and was passed into humans at the Huanan Seafood Market.
This “zoonotic” theory has no evidence to support its claim, and we’ll go through every single detail to understand why. The zoonotic theory argues that SARS-CoV-2 must be zoonotic because most other emerging infectious diseases are zoonotic, that two big lineages at the base of the tree could only be caused by two spillover events, and cases provided by China - a government well known to be untruthful on issues that affect its national security - cluster near both the wet market & the Wuhan CDC so therefore the virus must have come from the wet market.
The claim that most emerging infectious diseases are zoonotic therefore SARS-CoV-2 must be zoonotic is like saying that because most deaths are due to natural causes, the people who died in Chernobyl must also have died of natural causes, despite clear evidence suggesting otherwise. We must not let our priors determine our conclusions, as our priors may be wrong. We need to focus on the evidence that does and does not support claims.
Let’s dive into the lack of evidence for the zoonotic theory.
All viruses come from somewhere. Even a coronavirus that arises in the middle of Wuhan, walking distance from world-famous coronavirus research facilities, can trace a chain of causation back to a wildlife reservoir. The question before us is whether or not that chain of causation preceding SARS-CoV-2 includes clear causality by the activities of researchers.
Zoonotic viruses that are novel to humans must first persist in wildlife populations, and those wildlife populations must interact with humans, either directly through bushmeat consumption or transfer of bodily fluids (e.g. racoon dogs with sniffles, or bats peeing in your drink), or indirectly through vectors like mosquitoes or ticks. SARS coronaviruses are not vector borne, so we can narrow our examination by noting the zoonotic theory requires the progenitor of SARS-CoV-2 to have existed in some animal reservoir that came in contact with humans. The zoonotic origin theory relies specifically on airborne transmission of a generalist mammalian pathogen from an animal exclusively to people in the Huanan seafood market, and not to any other animals in the market. The reasons the zoonotic origin theory relies on airborne spillover, and not bushmeat consumption or direct contact (kissing a cat?), are (1) in the wet market, the virus is found over many surfaces across many rooms and (2) the zoonotic origin theory relies on superspreading in the market, and neither direct contact nor bushmeat consumption enable superspreading (viruses like HIV can exhibit superspreading, but over the course of years in hubs of sexual contact networks).
It’s hard to reconcile the necessary epidemiology at the heart of zoonotic origin - an airborne transmissible, generalist mammalian pathogen in a dense market full of animals - with the fact that the virus has not been found in any animals in the market besides humans, and not for a lack of effort. In just three months after the emergence of SARS-CoV-1 back in 2002, researchers sampled 25 animals, fewer animals than kids in a typical classroom, and found 7 animals tested positive with extremely close relatives of the virus found in humans. As 7 kids in a classroom can be sick with the flu, spillover commonly occurs during outbreaks and periods of high prevalence in animal populations, such as the current cases of H5N1 that are occurring during a global H5N1 outbreak in animals. Pathogens that spillover into a world with modern biotechnology and considerable knowledge of wildlife reservoirs should be easily discoverable by testing those animals in the exact market in which spillover is hypothesized to have occurred.
However, shortly after the Huanan seafood market outbreak, researchers (Gao et al.) sampled over 450 animals from the Huanan Seafood Market, over 15 times as many animals sampled for SARS1, and they found nothing. An intensive surveillance effort in the wet market shortly after the hypothesized multiple-spillovers in that exact market did not find a single animal that tested positive for a SARS coronavirus, let alone a close relative of SARS-CoV-2. If SARS-CoV-2 came from animals, specifically a highly infectious, superspreading animal spewing a virus capable of jumping the species barrier in two spillover-superspreading events, why can’t we find a single animal in the wet market infected with a cousin of SARS-CoV-2?
For other bat viruses, we can find reservoirs very quickly. After the recent outbreak of Nipah virus in Kerala, India, colleagues and I gave recommendations of which bats to test and, within months, researchers found a Nipah virus progenitor in bats. The same story goes for Swine flu in 2008, Hendra in Australia, and MERS-CoV emerging in June 2012 for which evidence of the virus in dromedary camels was found by June 2013. Should the current H5N1 outbreak cause a pandemic, we will have already documented the progenitor in birds every year since 1996. Spillover is not black magic: viruses come from somewhere, and for most viruses we have decades of experience sampling wildlife reservoirs & understanding when, where, and why they spillover. Our modern technology for DNA sequencing enables us to rapidly sample many animals for wildlife viruses, enabling swift detection of reservoirs not possible for the most elusive emerging infectious diseases of the 20th century like Ebola and HIV. However, thanks to advances in biotechnology, we have found reservoirs of both Ebola and HIV. We are standing on the shoulders of giants with our modern biotechnology and knowledge of animal reservoirs. From the genome of SARS-CoV-2 alone, we were able to look at other related viruses, look at peculiarities in its genetic code, and without sampling a single animal we could tell SARS-CoV-2 has some distant connection to bats. As a relative of SARS-CoV-1, we had reason to suspect civets, raccoon dogs, and other animals of the animal trade as possible intermediate hosts driving emergence. From Nipah and Hendra to SARS-CoV-1, MERS-CoV, zoonotic influenzas and more, our modern biotechnology, wildlife virology knowledge, and animal surveillance capacity allows us to find reservoirs for serious emerging infectious diseases in less than a year, especially in a place with the renowned wildlife SARS coronavirus surveillance capacity of Wuhan.
It’s been three years, and we’ve found nothing. Thousands of animals have been surveilled with our modern DNA sequencing and comprehensive knowledge of animal reservoirs, and we have found nothing. This is abnormal lack of reservoirs despite considerable effort is significant evidence against a zoonotic origin of SARS-CoV-2.
In addition to discoverable reservoirs, zoonoses usually leave clear footprints of infections at the human-animal interface. After the emergence of SARS-CoV-1, researchers weren’t sure where the virus was spilling over, so they studied the blood of animal handlers all around Guangdong province. They found that 58% of animal handlers who handled civets, a relative of the mongoose, had antibodies for SARS-CoV-1 in their blood, whereas animal handlers who handled snakes, birds, and fish had significantly lower levels of antibodies in their blood. Finding evidence of human infections concentrated in a very particular set of humans with very specific human-animal interactions scaffolded the zoonotic theory for SARS-CoV-1, giving us good reason to believe civets in animal trade networks were the reservoir. Indeed, sequence surveillance of animals later confirmed civets as an intermediate host driving the SARS-CoV-1 pandemic.
In contrast, Gao et al. sampled the surfaces near animal handlers and vegetable stalls in the Huanan Seafood Market and found no significant difference in the rate of positive samples (technically, vegetable stalls were nearly twice as likely to test positive, but the difference was not statistically significant due to small sample sizes). The absence of a higher concentration of viruses or infections around animals in cages suggests the virus was not lurking in an animal in a cage, but rather was spewed around the wet market by humans who walked around and visited animal and vegetable stalls alike. The Huanan seafood market yielded no reservoir, no evidence of infections in animal handlers, and no higher concentration of viruses around animal cages compared to vegetable stalls. The lack of critical zoonotic evidence that we searched for weighs heavily against the hypothesis that the Huanan seafood market was a site of spillover. We’ve looked and we’ve found nothing, leaving us with no evidence for a zoonotic origin of SARS-CoV-2. The missing zoonotic evidence strongly suggests the Huanan seafood market was not a site of spillover, but a site of transmission.
But what about those two papers widely reported to have “solved” this issue and provided “dispositive” evidence that SARS-CoV-2 spilled over twice in the Huanan Seafood Market? Those two papers made strong claims and conclusions that were not justified by their methods. There used to be scientists and papers aplenty confidently claiming the Earth is the center of our solar system and objects can travel faster than the speed of light, yet by using evidence and reason we can throw those old papers into the intellectual rubbish bin. History is full of scientists who were confident, prestigious, and wrong. We can use evidence and reason to see why the papers were fatally flawed, and later papers have shown us why. As Albert Einstein said, “It’s good to be first. It’s better to be right.”
When we look closely at those papers, we can see what’s wrong about them.
The first paper, Pekar et al., noted that the evolutionary tree of SARS-CoV-2 (shown below) contains two, large branches at the base of it, referred to as “basal polytomies”. At the bottom of the SARS-CoV-2 evolutionary tree, there is one cluster of branches, lineage A, that has many descendants radiating from the same common ancestor. There is another lineage, lineage B, that is descendant from Lineage A and also has many descendants radiating from a single common ancestor branching off lineage A.
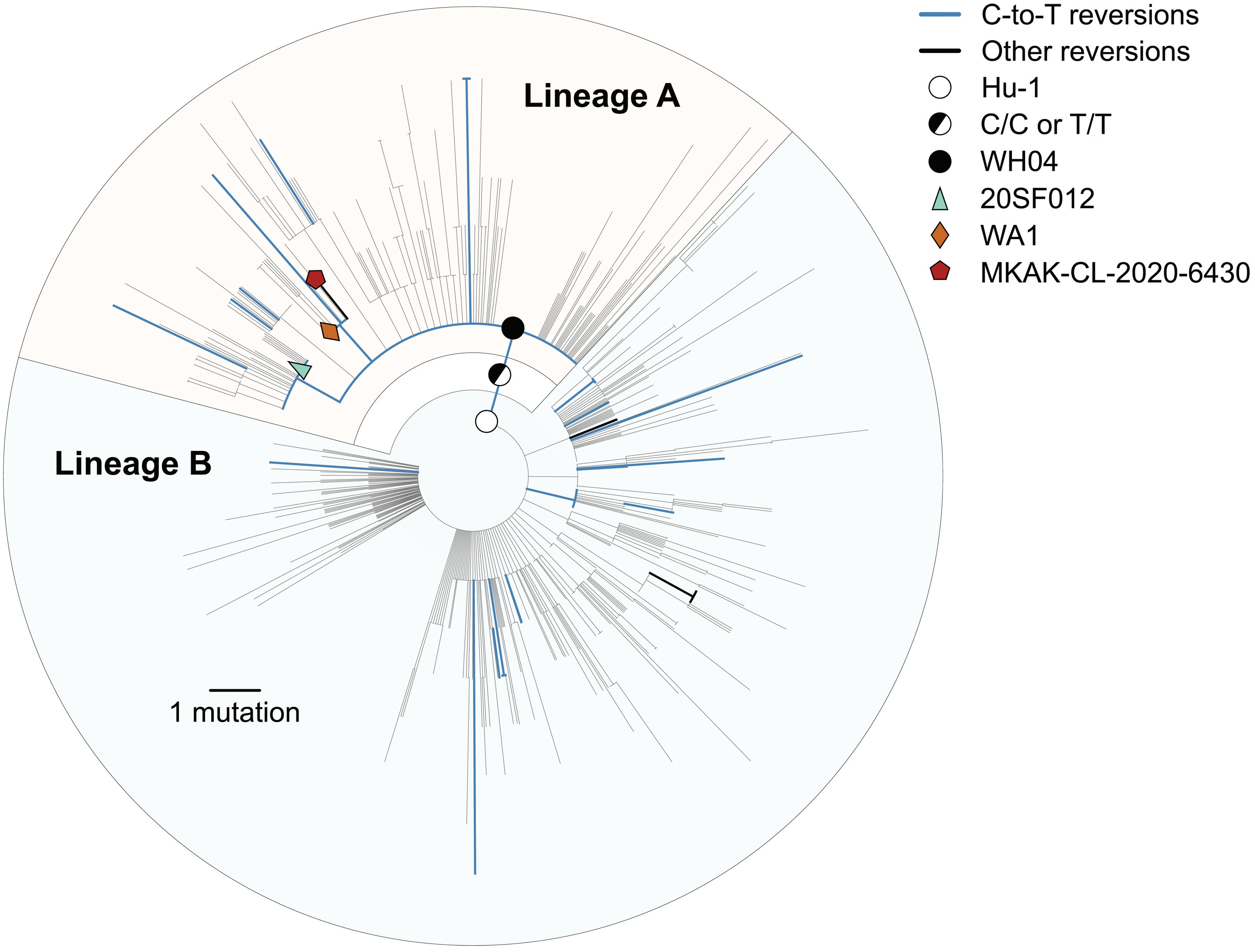
Pekar et al. tried to say that a tree with this structure is unlikely to occur by one spillover. To estimate the odds of seeing a tree like this one, the researchers used a toy model built for HIV, a sexually transmitted disease that causes chronic infections during which the virus evolves within hosts. Using the HIV model, Pekar et al. simulated outbreaks and computed the odds of seeing the SARS-CoV-2 evolutionary tree under their HIV-model of viral transmission and evolution. Clearly, HIV is not SARS-CoV-2. The researchers assumed Chinese authorities drew cases at random from the thousands of early cases. From this highly unrealistic toy HIV model of viral transmission, evolution, and case-ascertainment, the researchers estimated a low probability of two basal polytomies. The only other possibility, they claimed, was that the virus must have spilled over twice. However, they did not calculate the odds of seeing this exact tree under a scenario with two spillovers, nor did they justify how spillover - as opposed to superspreading - would produce a big polytomy in the first place.
Colleagues and I wrote a paper clarifying the fatal flaws of this work. Our results are simple and easy to understand.
SARS-CoV-2 is a superspreading virus. Superspreading events at choir practices or wet markets will create large lineages like lineage A and lineage B. The index patient will be infected with one strain, infect dozens of close contacts, and each close contact will then harbor a viral population that branches off the common ancestor in the index patient. Superspreading creates polytomies. While HIV is a superspreading virus, HIV superspreading occurs over long timescales during which the virus can evolve within a host, so HIV superspreading will often result in one patient passing on many different lineages as the virus evolves within that superspreading patient. SARS-CoV-2, on the other hand, causes superspreading events on the timescale of hours. Over 60 people can be infected with the same strain of virus in a densely packed market, creating a single, large lineage like those observed in the SARS-CoV-2 tree. The viral populations in each of those 60 people diverge like Darwin’s finches on different island of the Galapagos, and a polytomy in the SARS-CoV-2 tree blooms.
Pekar et al. further assume cases were drawn completely at random in the Wuhan population, yet we have incontrovertible evidence that authorities in China did not sample cases at random. Health authorities in China performed contact tracing, preferentially searching for patients with ties to locations visited by several of the index cases. Viruses in people who visited the same choir practice or wet market will likely be from the same lineage as those clusters of early ascertained cases. If you only sample patients in clusters, then you’ll only sample clusters of closely related viruses, and that can lead to large lineages. The unrealistic transmission & evolution assumptions combine with the failure to incorporate contact tracing to invalidate the conclusions of Pekar et al. They can’t claim the tree we observe is an unlikely event, because their model used to estimate its likelihood was unrealistic in several ways, all of which are well-documented empirical facts that make the tree we observed much, much more likely.
Speaking of empirical facts, it’s not even clear there are just two lineages at the base of the tree. Evolutionary trees are estimates of evolutionary history and, like any estimate, they are subject to change with more data. It turns out, there is more data. Pekar et al. excluded all sequences that conflicted with this story of two basal polytomies, and co-authors on our paper critiquing Pekar et al. found many sequences that likely yield intermediate branches between Lineage A and Lineage B. Additionally, Jesse Bloom uncovered sequences from the early outbreak in China that were deleted from NCBI, and those sequences may have branched off before Lineage A. Altogether, there are sequences excluded in Pekar et al. which change our estimate of the tree and completely undermine the empirical premise that there are two basal polytomies at all.
From the uncertain empirical premise to HIV superspreading models making unrealistic assumptions, we can’t put any stock in the strong conclusions claimed by Pekar et al. It’s more likely that human-human superspreading created the polytomies, contact tracing reinforced these polytomies, and other lineages exist but were not sampled well due to the limited and biased nature of early outbreak case ascertainment in Wuhan, and deletion of cases and sequences from the early Chinese outbreak.
We have to do one other sanity check of Pekar et al. The authors claim that the two lineages were caused by an animal transmitting the virus to a human twice in the same market. In order for these two spillover events to yield two polytomies, each animal—>human spillover event needed to be a superspreading event, otherwise the polytomy would have to have been formed by human-human superspreading and, if the polytomies were formed by human-human superspreading, then the polytomies provide no evidence of spillover. Let’s check-mate Pekar et al. by assuming there was a superspreading animal in the wet market and examining the logical implications of this hypothesis. It’s well documented that SARS-CoV-2 is a highly generalist mammalian pathogen - it can infect dogs, cows, deer, cats, people, tigers, bears, and more. If there were a superspreading animal with a highly transmissible, generalist mammalian pathogen in the wet market packed with animals, then that makes the discovery of 0 reservoirs out of 457 sampled animals even more anomalous to the point that it contradicts the assumption of a superspreading animal in the first place. If you assume a superspreading animal, you can’t explain the lack of infected animals in the market. There is a conflict between the model and assumptions of Pekar et al. and reliable data. It doesn’t matter how elegant or popular or hyped-up your theory is: if it conflicts with the data, then it is wrong.
We’re left with no choice but to conclude that there was no multi-superspreading non-human animal in the wet market, otherwise we would’ve at a minimum found infected animals in the wet market.
The other paper claiming to have provided “dispositive” evidence of a zoonotic origin is Worobey et al. The Chinese government provided locations of early cases to the researchers, the researchers looked at the locations of those cases scattered around Wuhan, estimated the “epicenter” of those cases, and found this epicenter was close to the Huanan Seafood Market. There are many problems with this, and they are easy to see.
First: can we trust the data provided by the Chinese government? The Chinese Communist Party can be counted on to be untruthful wherever the truth undermines their national security & self-interest, and a lab leak, if true, would be an historic embarrassment and national security threat to the Chinese government. The Chinese Communist Party lies. They lied about concentration camps of Uighur Muslims, military balloons floating over the United States, the militarization of the South China Sea, and more. The Chinese Communist Party has a clear conflict of interest given that the labs at the heart of a lab-leak theory are their own. If there were a lab leak, what kind of data would the Chinese government provide? As the saying goes, there are lies, damn lies, and statistics - it would be extremely easy for China to subsample data given to Worobey et al. in a way that ensures statistics tell the lie they wish to tell. The untruthfulness of the Chinese government and their clear conflict of interest means all data they provide has the risk of being intentionally misleading. We have to corroborate data from multiple sources before we can trust it.
Worobey et al. trusted this data from the Chinese government without question. Worobey et al. did not assess whether or not earlier cases, or cases providing some evidence of a lab origin, were excluded (foreshadowing: there are earlier cases and evidence of a lab origin that were excluded). With those credulous assumptions, Worobey et al. found the centroid of cases provided by the CCP was close to the Huanan Seafood Market. However, the centroid of these cases was even closer to the Wuhan CDC. If there were a lab leak in Wuhan, the Wuhan CDC would likely be involved in attempts to contain the early outbreak. It’s conceivable someone at the Wuhan CDC could get sick in their efforts & cause an outbreak in the same neighborhood as the Huanan Seafood Market. To believe the conclusions of Worobey et al., you must follow the footsteps of Worobey et al. by trusting Chinese authorities and the cases they provide, and by ignoring the proximity of the epicenter to the Wuhan CDC. You must also forget a key empirical piece of evidence where Gao et al. sampled animal & vegetable traders’ tables in the wet market, concluding the wet market was a site of transmission and contact tracing, not a site of spillover. Far from ‘dispositive’, Worobey et al. is unpalatably credulous despite the massive conflict of interest of the Chinese government, ease of manipulation, and independent lines of evidence conflicting with the data provided, including earlier cases preceding the wet market outbreak that were conveniently excluded from the analysis.
Some people point to the prestige of the journals, but everyone who has spent one day in science rolls their eyes at that. I don’t care if Worobey et al. were published by the Pope or Richard Feynman reincarnate, our job in science is to rigorously examine the data, methods, and logic of papers, and neither the data, nor the methods, nor the logic of Worobey et al. enables us to conclude the wet market was a site of spillover unless you just take the Chinese Communist Party’s word for it and ignore the existence of the Wuhan CDC and disregard the findings of Gao et al and so on. Our global jury is smarter than that. Did you take their word for it that Uighur concentration camps are just “re-education” facilities? That the balloon over Montana was for civilian purposes? That the islands in the South China Sea are for condos? Is our job as the jury to just take the “not-guilty” plea at face value? No. We have to skeptically examine every single claim, every point of data, and penetrate obfuscation to see the truth.
The common theme between Worobey et al. and Gao et al. is that there does appear to have been human superspreading events in the market. However, there are cases that precede the wet market outbreak of mid December 2019 and have no connection to it. There was a case reportedly from December 1 with no wet-market connection, and that event has independent corroboration through a spike in the usage of the keyword “SARS” on the Chinese social media app WeChat. When it comes to tracing the origins of an outbreak, a single earlier case carries more weight than a thousand later cases derived from a superspreading epicenter. The December 1 case was not in the Worobey et al. dataset. The wet-market “epicenter” is rejected by several pieces of evidence we can independently corroborate that precede the wet market outbreak and tell a different story. The substantial evidence of untruthfulness from the Chinese government, and the independently corroborated December 1 confirmed case and SARS spike on WeChat, inspires complete doubt in the conclusions of Worobey et al.
Science advances by using evidence and reason to find flaws in old papers, and testing the logical implications of theories from multiple angles. The existence of two old papers claiming the pandemic originated in the Huanan Seafood Market is not proof that the pandemic originated in the Huanan Seafood Market. Even 2,000 papers saying the Huanan Seafood Marker was the origin would not be proof. Evidence and reason help us understand that the papers’ data and methods cannot justify their conclusions, and there are hundreds of thousands of papers in the intellectual rubbish bin after being shredded by critical examination of their data, methods and logic. Pekar et al. used a bad model to estimate the odds of a tree that might not be the right tree. Worobey et al. used incomplete data provided by an untrustworthy government to estimate a midpoint somewhere between a wet market and the Wuhan CDC. Newer papers using evidence and reason like that provided here have shown us why the old papers aren’t convincing many smart scientists.
That, folks, is science.
The two empirical-seeming papers claiming to have evidence of a zoonotic origin are wrong. There are other papers and arguments, none of which are scientifically significant but some of which have received undue media attention. Briefly, we have to talk about a few other over-publicized arguments commonly made for the zoonotic theory. In the Proximal Origin paper, a paper prompted, edited, and pushed by heads of health science funding who had funded the labs in question, there are a few dubious claims. One claim is that computer simulations of receptor binding reveal suboptimal receptor binding, therefore SARS-CoV-2 could not have leaked from a lab. That’s absurd. The argument considers a straw man of lab-origin scenarios as labs study many “suboptimal” viruses, especially wildlife virology labs, and there’s no reason why a virus produced by a lab would be optimal in any way. Even a virus serially passaged may not exhibit “optimal” receptor binding in a computer simulation, as viruses evolve to optimize fitness over their entire life cycle and too-good of receptor binding may cause the virus to bind on too hard or bind on to the wrong tissues, reducing fitness. Another claim we need to discard is the claim that the furin cleavage site (which we’ll discuss in greater detail below) is out-of-frame, and that an out-of-frame insertion would be “illogical”. First, the insert may not be out-of-frame (see below). Second, an out-of-frame insertion would be logical if there were restriction sites making an out-of-frame insertion easier than an in-frame insertion. As surely as you can compl___ __is sentence, researchers can complete genomic sentences and cleverly insert sequences out-of-frame to create a desired virological product.
The conclusions presented in Worobey, Pekar, and Proximal Origin are not supported by their data, methods, and logic. Consequently, there is no stack of literature we can lean on to assume spillover occurred. We have to go back to the zoonotic evidence we’ve looked for but not found to appreciate the significance of the missing zoonotic evidence.
When SARS-CoV-2 emerged in Wuhan, walking distance from the world-class surveillance capacity of the Wuhan CDC and a world-leading coronavirus lab, with modern knowledge of wildlife reservoirs of SARS-CoVs, researchers looked for coronaviruses in animals and found nothing. We’ve looked for aliens on the moon and never found them - that doesn’t *prove* aliens don’t exist on the moon, but it is strong evidence against the theory that aliens live on the moon. Similarly, we have looked extremely hard for evidence of spillover, and we have found nothing. That, in itself, is strong evidence against a zoonotic origin because, under the zoonotic theory, we expect something similar to the first SARS outbreak. Under the alternative theory that SARS-CoV-2 emerged from a lab, we expected these efforts would find nothing. We would expect no animals at the wet market would test positive, that surfaces underneath animal traders would be just as likely to test positive as surfaces under vegetable traders, that the Chinese government would create a narrative of non-lab origin while withholding the databases of coronaviruses studied by their labs, and we expected that there would be fatal flaws with studies claiming otherwise.
We now have to turn to the labs and the researchers in our search for the origins of SARS-CoV-2. Prior to the COVID-19 pandemic, organizations like EcoHealth Alliance were collecting coronaviruses from bats for years. They sampled thousands of bats, reportedly finding hundreds of SARS coronaviruses around southeast Asia, and they sent their samples to the Wuhan Institute of Virology. Researchers at EcoHealth Alliance and the WIV claim to have not found the progenitor of SARS-CoV-2 in all of those bats and all of those coronaviruses. They did, however, have a database with hundreds of unpublished SARS coronavirus genomes, a database that would single-handedly double, triple, or possibly quadruple our understanding of the genetic range of wildlife SARS coronaviruses. If the zoonotic theory were true, the database at the WIV would immediately exonerate the labs in question. Yet, that database was taken offline in the fall of 2019, close to the time when the pandemic is thought to have begun. After 3 years and 18 million deaths, and despite intense international pressure and scrutiny demanding they release their database and lab notebooks, the researchers and the Chinese government all refuse to cooperate. Why? If a zoonotic theory were true, the dataset would single-handedly absolve those parties of guilt and disprove the lab leak theory! Why haven’t they shared it?
Without the dataset, we have to notice that the Wuhan Institute of Virology was at the scene of the crime, with hundreds of SARS coronaviruses in its possession, with a long track-record of making recombinant SARS coronaviruses with increased human infectivity in sub-par biosafety conditions. The coronavirus researchers at the WIV have no alibi as they won’t share lab notebooks revealing the research they were conducting at the time of SARS-CoV-2 emergence. They refuse to share databases of coronaviruses in their possession that would exonerate them if and only if they did not create the progenitor to SARS-CoV-2. Furthermore, the leading bat coronavirus researcher at the WIV claimed nobody was sick in the fall of 2019, yet US intelligence found that 3 coronavirus researchers at the WIV sought care for flu-like symptoms. Why are they being so untruthful about COVID-like illnesses requiring the patients seek hospital care? Why is the Chinese government not forcing the researchers to share their alibis and databases with the world to remove the cloud over China and their labs if - and only if - they were innocent?
Has there ever been an innocent person charged with murder who refused to share their alibi that disproves the charges and secures their innocence? No. Absolutely not. Now, there is not one murder, but 18 million deaths. If I were charged with a murder that took place at this exact time, would I not tell everyone that I was here with you all in this courtroom at the time of the murder? The refusal of EcoHealth Alliance and the Wuhan Institue of Virology to share their dataset of coronavirus genomes is evidence against their case. They have in their possession a dataset that could fit on a thumb drive, uploaded to Dropbox, emailed in the blink of an eye, a dataset that, if we were to believe their claims of a zoonotic origin and the innocence of coronavirus labs in Wuhan, would instantly prove their innocence.
The could share their database, but they choose not to.
We can only infer that the dataset contains damning information of their research activities. We can only infer that it hasn’t been shared because the dataset contains the progenitor of SARS-CoV-2. We can only infer that the progenitor to SARS-CoV-2 on their dataset lacks the critical features of bioengineering that make this SARS coronavirus so clearly different, so evidently engineered, when compared to all other SARS coronaviruses known to modern science. We can only infer that they were conducting the risky research on bat coronaviruses when SARS-CoV-2 emerged, and that a lab accident in their dilapidated facilities staffed by insufficiently trained workers caused a bat coronavirus to emerge in Wuhan and kill 18 million people worldwide.
The Lab-Leak Theory
There is no evidence supporting the theory that SARS-CoV-2 is zoonotic.
We’ve looked for reservoirs and found none. The geographic pattern of the outbreak lacks the fingerprint of an animal trade outbreak. The surfaces underneath animal traders in the wet market were just as likely to test positive with SARS-CoV-2 as surfaces underneath vegetable traders. The evolutionary tree of SARS-CoV-2 is consistent with human-to-human superspreading. The cases clustered around the wet market are also clustered around the Wuhan CDC, they were all provided by a government with the largest conflict of interest on this matter, and attempts to independently verify the data quickly yield earlier cases with no connection to the wet market. The most valuable data to disprove a lab leak - sharing the database of coronaviruses studied by the Wuhan Institute of Virology - is being deliberately withheld by the Chinese government and the lab from which the virus is thought to have emerged.
The theory that SARS-CoV-2 came from a lab can explain all of this very easily.
There is a reason why SARS-CoV-2 didn’t emerge over a large geographic extent like earlier animal trade outbreaks, a reason why animal traders and vegetable traders were equally associated with SARS-CoV-2, a reason why we’re not finding reservoirs for SARS 2 despite sampling over 10 times the number of animals as in SARS 1, and a reason why the Wuhan Institute of Virology is not sharing their data.
SARS-CoV-2 emerged in a lab.
In addition to the evidence we lack for a zoonotic origin, we have a stack of incontrovertible evidence painting a clear, consilient story of research activities on coronaviruses in Wuhan that would generate a virus exactly like SARS-CoV-2. We also have evidence of unsafe biosafety conditions and emergency laboratory anomalies at the Wuhan Institute of Virology prior to & during the time when SARS-CoV-2 emerged. We have evidence of untruthfulness of the Chinese government and researchers studying coronaviruses in Wuhan or in collaboration with the Wuhan Institute of Virology.
In stark contrast to the zoonotic theory, there is an abundance of evidence supporting a research-related origin in this crime scene of SARS-CoV-2 origins. When combined, the totality of consilient evidence pointing to a lab leak of SARS-CoV-2 becomes overwhelming in support of a research-related origin, helping us reject beyond reasonable doubt a zoonotic origin of SARS-CoV-2.
There are six main sets of evidence which combine to form a crystal clear, consilient picture that SARS-CoV-2 arose as a consequence of research-related activities:
Geographic evidence of the SARS-CoV-2 emergence
The furin cleavage site
The human-specific codons in the furin cleavage site
The restriction map of SARS-CoV-2 consistent with an infectious clone
DEFUSE proposal to insert human-specific furin cleavage site in a SARS-CoV infectious clone in Wuhan
The behavior of Chinese researchers and authorities connected to the Wuhan labs
No single piece of evidence is enough to disprove the zoonotic origin theory, but each piece of evidence is an anomaly under the zoonotic theory and easily explained in a theory that SARS-CoV-2 arose as a consequence of research similar to that proposed in DEFUSE. After understanding and examining this evidence, especially how these many pieces of evidence combine to be stronger than the sum of the parts, it becomes clear beyond reasonable doubt that SARS-CoV-2 emerged from a lab.
The geographic evidence
There are clear patterns of which organisms live where, including microorganisms like viruses. We call these patterns “biogeographic” patterns, the geography of where you find different living things. Whales are in the ocean, ostriches are in Africa, kangaroos are in Australia, polar bears are in the arctic, and so on. Organisms live in specific places because they have specific evolutionary histories, barriers to travel such as oceans or mountains (or cities like Wuhan), and contemporary ecological niches determined by their physiological requirements for survival and reproduction.
Viruses and their reservoirs also have clear biogeographic patterns in where they occur and where they spillover. Nipah virus spills over in India and the Nipah belt of Bangladesh where flying foxes serve as reservoirs and folk drink date palm sap that has been contaminated by the urine of flying foxes. Nipah will not spillover in Houston as there are neither flying foxes nor anyone drinking date palm sap contaminated by flying fox urine in Houston. Hendra virus spills over in Australia where the reservoir flying foxes infect horses that infect the handlers of horses. Hendra will not spillover in Buenos Aires. Ebola spills over in Sub-Saharan Africa where bats have also been found. Ebola will not spillover in Antarctica. As surely as you find kangaroos in Australia and not America, Andean condors in South America and not Africa, bison North America and not Antarctica, and giraffes in Africa and not Asia, viruses have regular biogeographic patterns of where they’re found, where they spillover, and where they don’t.
SARS coronaviruses are not found in wildlife in Wuhan. The hotspot of wildlife SARS coronaviruses is in Laos and Malaysia, 1,000 miles away from Wuhan, or about the distance from New York City to Florida. You find alligators in Florida, not in New York City. Much like New York City, there are not a lot of bats or other wildlife in Wuhan. Wuhan is a densely populated, urban center with few places for bats to sleep and little for them to eat. Consequently, the zoonotic theory relies entirely on animal trade networks bringing animals with a coronavirus from Laos (or Yunnan province in SE China) to Wuhan. Yet, even the animal markets of Wuhan are not the largest nor the only animal markets in SE Asia. In fact, the map below shows where wildlife coronaviruses are likely to occur - Wuhan is in a desert of wildlife SARS coronavirus diversity. The entire range of wildlife SARS coronaviruses encompasses a population of 900 million people, only 11 million of which live in Wuhan. If you drew a person overlapping with a bat at random from this map, there’s less than 1% chance that this person lives in Wuhan, a global hotspot of wildlife coronavirus laboratory research.

Researchers sampled hundreds of animals in the animal trade network of Wuhan and didn’t find a single SARS coronavirus in any of them. Despite looking exactly where we should find evidence, we have found zero evidence of a wildlife coronavirus being in Wuhan. The distance of Wuhan from wildlife coronavirus hotspots combines with lack of any SARS coronaviruses found in extensive surveys of animals at the wet market to weigh strongly against the hypothesis of an animal trade outbreak.
The geographic evidence for a lab origin is strong evidence that should immediately compel transparency from Wuhan labs. Animal trade networks are geographically expansive, spanning the 1,000 miles connecting Laos to Wuhan and connecting a massive set of markets and animals, often stored in cages close enough together that the animals can expose one-another to whatever viruses they have. We would expect a SARS coronavirus circulating amongst animals in animal trade networks to infect a wide range of animals throughout this vast network of animal trade and animal traders, leading to high prevalence of the virus in animals and causing outbreaks in animal traders beyond Wuhan. Indeed, the animal trade outbreak of SARS-CoV-1 produced not one isolated outbreak walking distance from coronavirus labs, but many spillover events concentrated in animal traders over a vast geographic range spanning Guangdong province, a province about twice the size of Delaware, Connecticut, New Jersey, New Hampshire, Vermont, and Maryland combined.
That is what an animal trade outbreak looks like.
That is not what SARS-CoV-2 emergence looks like.
Prior to the COVID-19 pandemic, there had been 7 recorded SARS coronavirus outbreaks, including SARS-CoV-1. While SARS-CoV-1 provided a very clear and consilient picture of an animal trade outbreak across many lines of evidence, the other 6 out of 7 outbreaks were caused by laboratory accidents, most of them in China. The laboratory outbreaks of SARS coronaviruses all lacked the geographic trail of the SARS-CoV-1 animal trade outbreak; they caused singular outbreaks far from wildlife hotspots and in cities like Beijing, right next to labs studying coronaviruses. SARS-CoV-2 emerged in Wuhan and nowhere else, next to one of the largest coronavirus research labs in the world, leaving no geographic trail of infections consistent with an animal trade outbreak and emerging in a country where the majority of recorded SARS outbreaks have been lab-related.
The geographic evidence, and the historical context of previous SARS coronavirus outbreaks, provide a strong reason to believe that SARS-CoV-2 emerged in a lab.
The Genomic Evidence - a Primer
Next, let’s consider the genomic evidence.
The “genome” is the full set of genetic material encoding an organism. SARS coronavirus genomes are 30,000 A’s, U’s, G’s and C’s (‘base-pairs’) long. The SARS-CoV-2 genome is an “RNA” genome, which is similar to DNA and contains U’s instead of T’s in its genetic code. Genomes mutate over evolutionary time, sometimes turning an A to a U or a C to a G, and sometimes - but not frequently - by suddenly acquiring or deleting larger sequences. Modern biologists decipher genomes and study how the letters change from virus to virus in order to understand how viruses typically evolve. We also know the common tools and techniques for genetic engineering, and the fingerprints modern bioengineering efforts can leave in the genomes of genetically modified organisms.
This section will inevitably get a little more technical. Bear with me, though, because the technicalities are critically important. Let me give you an analogy. Below is a picture that is completely uninterpretable to a lay audience. It is a bunch of dots on a page arranged in rows and concentric circles, with a shadier circle of scattered points overlaid and a random white line cutting across half the page. What does this mean?

What you’re seeing in this picture is incontrovertible evidence, yet understanding the weight of that evidence requires technical expertise. In Jargon, the picture above is the X-ray diffraction pattern of a crystallized enzyme. In English, researchers shot X-ray beams at a crystal. Each dot is a place where the X-rays landed like bullets in a building after being deflected off a tank. Scientists measure how those beams bounce off small molecules arranged in the regular, gridded structure of the crystal, and use tried and true methods to translate the pattern above into 3-dimensional structures of the molecules in the crystal, a molecule our eyes can never see. Science provides the context to turn incomprehensible gibberish from complex measurements into incontrovertible evidence about our universe. As with X-ray diffractions, scientists can look at genomes and, while they use technical tools that take years to learn all the in’s and out’s, knowledge of these concepts and methods can help one readily identify patterns of the genetic code that are consistent with natural evolution of viruses, and patterns that are more consistent with genetically modified organisms.
The only way to evaluate the evidence of this case is for all of us to learn some biology.
By the end of this section on genomic evidence, we should all understand the significance of the genomic evidence and why it strongly suggests SARS-CoV-2 is a laboratory product, not a product of natural evolution. As immediately as you can see the fingerprint below, know it’s a fingerprint, and know the significance of such a print found on a gun, we all need to reach a level of fluency with molecular biology to recognize the furin cleavage site, the CGG-CGG encoding for arginine, and the BsaI/BsmBI maps, and understand the significance of these three independent pieces of evidence all indicating that SARS-CoV-2 was made in a lab.

The Furin Cleavage Site (FCS)
From the very first SARS-CoV-2 genome published from Wuhan, its genome was not like other SARS coronaviruses. In the middle of the Spike protein, there was a 12 nucleotide insertion referred to as the “Furin Cleavage Site” or FCS for short. The DNA sequence for this site was:
R1—CCU-CGG-CGG-GCA—R2
where R1 and R2 indicate the rest of the SARS-COV-2 genome on either side of the FCS (in other words, ignore the R1 and R2, let’s focus on the letters in the middle). Most of the SARS genome differs from close relatives by single mutations - an A where a close relative has a G, a C where a close relative has an A, and so on. The entire 12-nucleotide Furin cleavage site in SARS-CoV-2, however, is not found in any other SARS coronavirus.
Where did this insertion come from?
Furin cleavage sites were subjects of niche virological interest prior to COVID-19 because they are found in other viruses, including other very distant coronaviruses like MERS-CoV. A virus entering a cell has to do a sort of lock-and-key handshake with the host cell, and human cells have different ‘locks’ than bat cells. Furin cleavage sites, however, can make it much easier for viruses to “unlock” human cells. In Jargon, furin cleavage sites can facilitate receptor binding and reduce the barrier to entry, one of the main molecular biological hurdles that often prevents pathogens from entering a new host. Because the FCS is found in some viruses, and because it allow a virus to unlock human cells, researchers wondered in 2018: what would happen if we put a furin cleavage site in a bat SARS-CoV? Would it be better able to infect human cells?
Prior to SARS-CoV-2, a furin cleavage site had never before been observed in a SARS coronavirus. We had sampled hundreds of SARS CoVs around SE Asia and found nothing. When we reconstruct the SARS-CoV evolutionary tree, we can see over 1,000 years of evolutionary time where lineages branched off from one-another and, at every point in evolutionary time, they had every opportunity to acquire a furin cleavage site. Yet, they did not, at least not in all ~80 SARS CoVs we had discovered prior to the COVID-19 pandemic. In that entire millennium of SARS CoV evolution, there is not a single furin cleavage site, except for that found in Wuhan 2 years after researchers proposed to insert a furin cleavage site in Wuhan.
Let me repeat that slowly and clearly.
In over 1,000 years of evolutionary time, we see no evidence of a furin cleavage site in any SARS CoV, except for the SARS-CoV that emerged in Wuhan 1.5 years after researchers proposed to insert a furin cleavage site in a SARS CoV in Wuhan.
The Codon Bias of the Furin Cleavage Site
Take another look at the furin cleavage site:
R1—CCU-CGG-CGG-GCA—R2
I broke the nucleotides into sets of threes because the genetic code uses these triplets of nucleotides called “codons” to translate the genetic code from nucleic acids into amino acids. In our cells, DNA doesn’t do a lot of the heavy lifting - it doesn’t detect light, it doesn’t bring oxygen from the lungs to the legs when you run, it doesn’t breakdown your food, etc. Most of the core metabolic/physiological functions of our body are done by proteins, and proteins - like the Spike protein in SARS-CoV-2 - are made by chains of amino acids. DNA carries information to make proteins as organisms have ways to “translate” chains of nucleic acids into chains of amino acids, and those chains of amino acids form proteins like hemoglobin, keratin, and other proteins that enable living organisms to do what they do. SARS-CoV-2 RNA encodes a spike protein that binds onto a human protein (ACE2) encoded by our DNA. Nucleic acids translate to amino acids, and chains of amino acids ball up into “proteins” that do stuff. That’s the TLDR of how life works.
There is some redundancy in the genetic code. There are 4 base pairs in RNA (A, U, G, C) which produce 64 distinct triplets/codons, yet there are only 20 different amino acids. Since there are more codons than amino acids, multiple codons can encode for the same amino acid. Below is “the genetic code” showing the amino acid (Arg, Phe, Leu, Tyr, etc.) produced by every single codon. This genetic code is shared among humans, bats, viruses, plants, fungi, bacteria - every organism we know of uses this same genetic code. However, not every organism uses codons in the same frequency.
 | Khan Academy")
Notice that there are 6 different codons that can all produce Arginine: CGU, CGC, CGA, AGA, AGG, and CGG. Organisms will have what we call “codon biases” - we tend to prefer some codons and not others in a non-random way. When a virus evolves to a host, there is believed to be strong selection for the virus to use codons with similar biases as the host. For this reason, when SARS coronaviruses encode arginine, they tend to use AGA, AGG, or CGU. The rarest codon for arginine in SARS CoVs is CGG. It is used less than 1.5% of the time a SARS COV makes arginine.
When bioengineers are trying to move a genetic sequence from one organism into another, they think about codon biases. For example, these researchers wanted to put a jellyfish gene inside fungus so that the fungus would glow green. That’s cool. By switching the codons from the jellyfish codon bias to the fungal codon bias, researchers were able to ensure the recombinant fungus made more of the protein when this gene was being expressed. Codon biases are considered when making an RNA vaccine - vaccine makers may want to make Spike proteins in human cells to train the immune system to recognize Spike proteins on SARS-CoV-2. If they change the codons to be more human-optimized, they may be able to make a more effective vaccine thanks to cells producing more Spike protein with less RNA.
If a researcher wanted to insert a novel furin cleavage site inside bat SARS-CoV and test its ability to infect humans, they would very likely use “human-specific” codons.
When humans make arginine, we tend to use the codon CGG more than any other arginine codon. In other words, the furin cleavage site sequence below:
R1—CCU-CGG-CGG-GCA—R2
is “human-specific” in that it uses codons that are expected in a human and exceedingly rare in bats and SARS CoVs. The human-optimization of the furin cleavage site is additional evidence of a lab-origin of SARS-CoV-2 given the common practice of codon-optimization in bioengineering. As there’s less than a 1/1,000 chance of seeing a furin cleavage site appear in SARS-CoV any given year,, there is a roughly 1/400 chance of seeing 2 CGG codons in the same inserted sequence if this insert had the same codon bias as the rest of the SARS-CoV-2 genome. Given selective pressures on a bat CoV to mimic the codon biases of its host, it’s unlikely this FCS existed in a bat CoV, otherwise we’d strongly expect this insertion to use different codons for arginine.
There are other explanations. Maybe the FCS was acquired by recombination with an animal (e.g. a pangolin) that also had a human-like CGG preference for arginine. The codon bias alone does not imply that the FCS is engineered. It does, however, add to the stack of evidence that is more easily explained by a lab origin than a natural origin, especially given the nature of research proposed on CoVs in Wuhan.
If someone were trying to make an FCS for humans and familiar with the common literature on codon-optimization, they’re likely to choose the most common arginine codon for humans twice in a row.
In 2018, researchers proposed to insert a “human-specific” furin cleavage site in a SARS-CoV in Wuhan.
In 2019, SARS-CoV-2 emerged with a “human-specific” furin cleavage site in Wuhan.
Restriction Map of an Infectious Clone
In addition to the human-specific codons in the furin cleavage site, the SARS coronavirus in Wuhan has another feature in its genome that is consistent with lab-made coronaviruses and anomalous in nature.
In order to make a coronavirus in a lab, researchers have to build its genome one piece at a time. That requires using special molecular “cutting and pasting” methods to cut up lego-blocks of DNA and paste them together in the right order. Researchers would typically paste together a full-length (30,000 base-pair) DNA ‘clone’ of a coronavirus, transcribe that DNA into RNA, and then insert the RNA into a cell. The cell would treat that RNA just like any other viral RNA: it would start making proteins, those proteins would make a virus, and boom: a viral “infectious clone” would be born by the immaculate conception of modern biotechnology.
Commonly used pre-COVID methods to make infectious clones left a fingerprint in the genome. Researchers typically wanted to build the clone using as few different molecular “scissors” or “restriction enzymes” as possible, yet no single chunk of DNA could be too long so they had to keep the longest fragment of DNA as small as possible. Restriction enzymes cut DNA at very well-defined sequence called “restriction sites” and we can see where those restriction sites are in the genome. Researchers would often look at the genome, find the restrictions sites, and then add + remove restriction sites with “silent mutations” that change the DNA sequence to new codons that encode for the same amino acid but change a restriction site (e.g. changing CGG to CGU, to encode arginine with a different codon but let scissors that recognize “CGU” cut the genome at this site). The process of adding and removing restriction sites to make infectious clones allowed researchers to cut the viral genome into as few chunks as possible while keeping the maximum fragment length small.
Two of the most popular scissors for this job - BsaI and BsmBI - were used around the world and were available to the WIV prior to COVID-19 as evidenced by their prior work utilizing these enzymes.
When we look at the BsaI + BsmBI restriction sites of SARS-CoV-2, we can see that cutting up the genome with these enzymes would produce the ideal number of cuts proposed for these ‘reverse genetic systems’ for infectious clones, and the length of the longest fragment is significantly shorter than expected by chance. In this regard, the genome of SARS-CoV-2 is like an Ikea virus - it’s ready to assemble from the get-go - and almost no wild viruses meet this criteria. The restriction map of SARS-CoV-2 is a rarity among wild coronaviruses, yet it is exactly what we would expect from an infectious clone. We estimated <1/1,400 chance of seeing a restriction map as-or-more optimized for infectious cloning than SARS-CoV-2.

That’s not all.
In addition to having a pattern of cutting/pasting sites in its genome that makes SARS-CoV-2 a rarity in nature and look more like an infectious clone than several documented infectious clones, the mutations that differentiate these restriction sites from close relatives are all the “silent” mutations researchers would use to add and remove restriction sites to achieve this “Ikea virus” design.
What’s more, there is a significantly higher concentration of silent mutations within these restriction sites than in the rest of the genome (P=5e-8 for RaTG13), and evolutionary simulations suggest it is highly unlikely one could obtain such a significant infectious-clone restriction map from natural evolution of the closest relatives of SARS-CoV-2. While this restriction map is a rarity in nature that is extremely unlikely to occur by natural evolution, it is exactly what we would expect from a product of pre-COVID research on coronavirus infectious clones.
To summarize, SARS-CoV-2 has a BsaI/BsmBI restriction map consistent with an infectious clone, and meets all the criteria of an infectious clone including the shortened longest-fragment-lengths, restriction sites produced exclusively by silent mutations, and a significantly higher concentration of silent mutations within these sites than the rest of the genome. SARS-CoV-2 looks a lot like an infectious clone, much more than the vast majority of wild coronaviruses.
In 2018, researchers proposed to insert a human-specific furin cleavage site in a SARS-CoV infectious clone in Wuhan.
In 2019, SARS-CoV-2 emerged in Wuhan with a human-specific furin cleavage site and the restriction map of an infectious clone.
The DEFUSE proposal
We’ve gone over the geographic and genomic evidence that makes SARS-CoV-2 highly unusual among other SARS coronaviruses. SARS-CoV-2 emerged in Wuhan in 2019 far from the hotspots of wildlife coronavirus diversity, right next door to world leading labs studying wildlife coronaviruses, and it emerged with a human-specific furin cleavage site and the restriction map of an infectious clone.
Less than 1.5 years earlier, researchers at the Wuhan Institute of Virology and elsewhere proposed to insert a human-specific furin cleavage site in a SARS coronavirus in Wuhan.
Read those last two paragraphs again.
The DEFUSE proposal is where researchers laid out their intentions to make a virus shockingly similar to SARS-CoV-2 in all of the ways in which SARS-CoV-2 is glaringly different from wildlife SARS coronaviruses.
The DEFUSE proposal was a grant proposal written by Peter Daszak at EcoHealth Alliance in NYC (EHA), Zheng-Li Shi at the Wuhan Institute of Virology (WIV), Ralph Baric at the University of North Carolina (UNC), Linfa Wang at Duke-NUS Singapore, and others. The proposal was submitted to DARPA’s PREEMPT call, a grant call looking for innovative ideas to preempt pathogen spillover before it occurs.
The DEFUSE proposal contained many specific aims: catching bats, sending samples from bats to labs, looking for viruses in the samples, studying and modifying the viruses in labs, developing raccoon poxvirus vaccines to boost immunity & protect bats against the viruses, testing the viruses + immune-boosting in bats, forecasting where spillover is most likely to occur, and deploying vaccines in wild bats to preempt spillover.
If you look on page 11 of the main document, page 13 of the online PDF above, under the section labelled “S2 proteolytic cleavage and glycosylation sites”, you can find the most important passage in this document.
The researchers propose to scan SARS-CoV genomes for potential furin cleavage sites and, where none exist, they propose to insert the appropriate cleavage sites. Specifically, they say:
”… we will introduce appropriate human-specific cleavage sites and evaluate growth potential in Vero and HAE cells”
In other words, a major aim of the DEFUSE proposal was to study furin cleavage sites, features never before documented in SARS CoVs. They would look for furin cleavage sites that might exist and, where none existed, they will introduce human-specific cleavage sites to create a virus not found in nature but hypothesized- for good mechanistic reasons based on our knowledge of furin cleavage sites - to have higher transmissibility in people and, consequently, a higher risk of causing a pandemic like the COVID-19 pandemic. Where none existed, researchers proposed to insert human-specific furin cleavage sites in viruses, and study whether or not their poxvirus vaccine protects bats from these unnatural coronaviruses they designed. Studies of the infectivity of high-risk strains in bats, and whether or not the immune boosting methods at Duke-NUS protected bats from high-risk strains, was proposed to take place at the Wuhan Institute of Virology.
The DEFUSE grant was not funded, but that doesn’t mean the research didn’t continue. Scientists are often like actors: the show must go on. EcoHealth Alliance had many sources of funding, and the proposed research inserting a human-specific FCS in a bat SARS-CoV is relatively inexpensive. In fact, three PIs of DEFUSE - Peter Daszak, Lin-Fa Wang, and Zheng-Li Shi, had created a chimeric bat coronavirus in 2016 without the involvement of UNC. Peter Daszak had a grant at NIAID - Understanding the Risk of Bat Coronavirus Emergence - that funded a collaboration between EcoHealth Alliance and Chinese researchers, including Zheng-Li Shi at the Wuhan Institute of Virology, to study bat coronaviruses in China (Ralph Baric was missing from both the 2016 paper and, apparently, the NIAID grant). The NIAID grant was listed in the acknowledgements for their construction of a recombinant bat SARS CoV in the 2016 paper. Researchers in China also had access to alternative lines of funding. The researchers had ample means to follow through with their intentions spelled out in DEFUSE, especially relatively inexpensive work but exciting work such as inserting human-specific furin cleavage sites in SARS CoV infectious clones.
In 2018, researchers indisputably proposed to insert human-specific furin cleavage sites in bat SARS-CoVs in a collaboration involving the Wuhan Institute of Virology. Their prior work creating recombinant CoVs used a particular method to construct infectious clones of viruses using reverse genetics systems.
In late 2019, SARS-CoV-2 emerged with a human-specific furin cleavage site in what otherwise looked like a bat SARS-CoV right outside the Wuhan Institute of Virology. This virus is anomalous among wild coronaviruses in just how consistent it is with reverse genetic systems, all the way down to silent mutations significantly concentrated in restriction sites.
Every single anomalous feature of SARS-CoV-2 that leads us to suspect a lab origin, features not seen in over 1,000 years of evolutionary time, was spelled out in a grant just over 1 year prior to the emergence of the virus. That grant did not propose to do its work in Atlanta nor Athens nor Cape Town nor Milan nor Buenos Aires. It proposed to do this work in Wuhan.
The virus with this lab-looking genome was not found in animals in the wet market. It was equally likely to be found underneath animal traders as vegetable traders. It did not cause a geographically widespread outbreak consistent with an animal trade outbreak. Simply put, it did not look zoonotic in ways that are easily explained by a lab origin, and it has a genome that looks exactly like a research product from a proposal to make recombinant bat coronaviruses in Wuhan.
Behavior of coronavirus researchers before & after SARS-CoV-2 emergence
If a zoonotic origin were true, the labs in question would know it and the lab origin theory of SARS-CoV-2 could’ve been disproven by simple transparency from the labs in Wuhan. However, rather than allow international teams to visit their labs, rather than provide serological evidence showing no signs of SARS coronavirus infections in laboratory workers, rather than release their dataset of coronaviruses being studied in Wuhan, the Chinese government has blocked access to its labs, limited speech on COVID-19 from its scientists, provided incomplete data to Worobey et al., deleted sequences from servers that shine critical light on early SARS-CoV-2 evolution, and changed its narrative about viral origins erratically, first saying it came from a wet market, then saying it arrived in frozen meat, and now saying it emerged in USA despite the earliest outbreak indisputably occurring in Wuhan, as evidenced by early reporting & hospitalization surges and traveler screening of passengers flying out of Wuhan in early 2020. Despite having the most extensive database of SARS-CoVs in the world at the WIV, the Wuhan Institute of Virology and the Chinese Communist Party refused to release their dataset of coronaviruses or allow independent teams to examine their coronavirus research labs. If the Chinese Communist Party believed its narrative that SARS-CoV-2 emerged in the USA, why not clear its name by sharing data and letting international teams inspect their labs?
Zheng-Li Shi claimed that nobody was sick in the Wuhan Institute of Virology in the fall of 2019, yet there were many major events in the institute that contradict her claims. In September 2019, the Wuhan Institute of Virology took its database of SARS CoVs offline, around the same time the institute switched from civilian to military control, a contractor was hired to fix HEPA filters in the lab where broken HEPA filters can cause lab-acquired infections (uh oh), and China began stockpiling personal protective equipment at an unprecedented scale. In October 2019, cell phone location data reveal a complete shut-down of the lab. By November of 2019, US intelligence agencies had evidence of a massive outbreak in China occurring (intelligence that precedes the earliest cases provided to Worobey et al.), and, at some undisclosed time in the fall of 2019, 3 coronavirus researchers at the Wuhan Institute of Virology sought care for severe, influenza-like symptoms, symptoms consistent with COVID.
While the Chinese government says the first cases were detected in late December, US intelligence and other alternative data streams reveal clear evidence of a major epidemiological event starting as early as September 2019, with highly relevant epidemiological anomalies continuing throughout the fall until, finally, the Chinese government reported a novel coronavirus causing a “pneumonia of unknown etiology” in early January 2020. While Zheng-Li Shi says that no workers at the WIV fell ill in the fall of 2019, US intelligence agencies have evidence that 3 workers became sufficiently sick that they all sought care. Why is the Chinese government being so untruthful, and why are they still not sharing the database of CoV genomes they took offline in September 2019?
Because SARS-CoV-2 leaked from a Chinese lab, the truth would reveal the Chinese government’s role in covering up the earliest cases in the worst pandemic of the past century, and the database contains the progenitor used to make SARS-CoV-2.
Meanwhile, Peter Daszak, the president of EcoHealth Alliance, has obstructed investigations and obfuscated the facts of the matter. In March 2020, Peter Daszak wrote a letter to The Lancet calling all non-natural theories of SARS-CoV-2 origins “conspiracy theories”, without disclosing the conflict of interest that he studied bat SARS coronaviruses with the Wuhan Institute of Virology for years, had several active grants, papers, and other collaborations ongoing with the Wuhan Institute of Virology, and even wrote a proposal in 2018 to make a virus, in Wuhan, that looked exactly like SARS-CoV-2.
FOIA’d emails obtained by US Right to Know reveal Daszak’s thought process behind the letter, indicating a clear intent to deceive the public, to use The Lancet to manipulate public opinion and thereby deflect attention away from his collaboration. Writing to Ralph Baric, a co-PI on the DEFUSE grant not listed as an author on the Lancet Letter, Daszak says:
“I spoke with Linfa [Linfa Wang, Duke-NUS co-PI of DEFUSE] last night about the statement we sent round. He thinks, and I agree with him, that you, me, and him should not sign this statement so that it has some distance from us and does not work in a counterproductive way.
… We will then put it in a way that doesn’t link back to our collaboration so we maximize an independent voice.”
From this email, Daszak expresses his clear intentions to produce a letter “maximizing a public voice” and calling lab-origins a “conspiracy theory” while offering to remove the three top suspects in the lab-origin investigation from co-authors so it doesn’t “link back to our collaboration” and “has some distance from us and does not work in a counterproductive way”. Daszak clearly sought to deceive the readers of the letter, to manipulate public and scientific opinions on the matter to deflect attention from his group and to do so in a way that did not trace back to him and two other co-authors of the DEFUSE grant proposing to make a virus just like SARS-CoV-2 in a lab in Wuhan.
Peter Daszak was appointed to lead of the Lancet Committee investigating COVID-19 origins. Again, Daszak did not disclose his massive conflicts of interest. When the chair of the broader Lancet Commission Dr. Jeffery Sachs found out about Daszak’s COI and similar undisclosed COIs of researchers recruited to the taskforce by Daszak, Dr. Sachs shut down Lancet’s investigations on COVID origins. By failing to disclose his COIs while occupying the top post on the committee, and by appointing people who had similar COIs, Daszak successfully obstructed The Lancet’s investigation into SARS-CoV-2 origins, preventing scientists from receiving an honest investigation from a leading, globally trusted medical journal that clearly desired to uncover the facts.
The World Health Organization sent a team to investigate the origins of COVID-19. Peter Daszak asserted himself as the emissary for the United States in this effort and, again, did not disclose his conflicts of interest. Although we don’t know what role Daszak played in his capacity as US emissary, the WHO team turned over no evidence, and we are left wondering if he influenced the investigation in a manner that prevented the WHO team’s ability to uncover facts that may incriminate Daszak or his colleagues in the creation of SARS-CoV-2. Daszak managed to publish papers in The Lancet calling lab-origin theories “conspiracy theories and repeatedly inserted himself into key nodes of scientific and investigative power to oversee and torpedo investigations into his own proposed research activities in Wuhan.
Why so much manipulation, deception, undisclosed COIs, and obstruction?
The DEFUSE grant itself is a critical piece of evidence on SARS-CoV-2 origins. It is the recipe book proposing to make a virus exactly like SARS-CoV-2 in Wuhan, yet this essential piece of evidence was not shared by Peter Daszak at any point in time, least of all when he was on committees investigating a possible lab origin, committees that would’ve been greatly interested in the evidence Daszak held. Rather, the DEFUSE grant was obtained against the will of Daszak and released by online sleuths.
The untruthful and manipulative behavior of researchers before and after the first internationally reported cases indicate a consciousness of guilt and an effort to obstruct investigations into their own research. The Chinese government is untruthful because it knows it is responsible for pushing risky research in sub-par biosafety research facilities, facilities that needed new HEPA filters suggesting they were in disrepair & prone to lab accidents. Zheng-Li Shi was untruthful about whether or not anyone was sick in the WIV because she knew the truth - that there was an outbreak of influenza-like illness among her community of coronavirus researchers - would naturally lead to questions about the nature of their research and uncover the lab origins of SARS-CoV-2. Peter Daszak withheld DEFUSE, did not disclose his COIs, manipulated public opinion, and obstructed investigations because he knows that his own organization supplied the Wuhan Institute of Virology with a bat virus from SE Asia, collaborated with the Wuhan Institute of Virology to make recombinant bat coronaviruses, and proposed a grant that serves as a perfect blueprint to make SARS-CoV-2. Daszak understands the significance, and the consequences, of a virus exactly like the one they proposed to make and study at the Wuhan Institute of Virology emerging to kill 18 million people worldwide. That’s why China is not truthful about incidents at the WIV and early cases, that’s why China refuses to cooperate with international investigations in a lab origin, that’s why Zheng-Li lied and Daszak obstructed global scientific and health investigations into the origins of SARS-CoV-2: they created the virus, they are conscious of their guilt.
Closing the case
It’s tragically ironic that DEFUSE was submitted to the “PREEMPT” program. I was a mathematical biologist working with a PREEMPT team to forecast pathogen spillover, so this whole odyssey is pretty close to home for me. The subject matter expertise I gained on pathogen spillover has helped me identify the zoonotic evidence we lack, and my unique role statistical analyses and forecasting spillover provides a unique perspective on the significance of the evidence indicating a lab origin. Forecasting is helpful because it helps us prepare, and also helps us consider alternative realities in which we prevent an undesirable outcome like a pandemic. How might we have preempted the COVID-19 pandemic?
Let’s go back in time and imagine the crime scene from the 2 years before the pandemic. If you were me in 2018, forecasting spillover, and asked to guess where a SARS CoV pandemic would emerge and what the viral genome would look like, what would you predict? How would you stop the oncoming pandemic?
You would look at bat-human overlap in SE Asia and estimate Wuhan has a <1% chance of being the site of spillover. There was never, by that time, a furin cleavage site documented on the SARS-CoV evolutionary tree, so you might guess there isn’t going to be an FCS at all. However, if you were forced to assume an FCS is possible, you might estimate the rate at which FCS’s are inserted in the SARS-CoV tree, assume 1 shows up in the 1,000 years of evolutionary time, and so you’d give it less than a 0.1% chance of occurring. If someone told you there were two arginines in the furin cleavage site (i.e. conditioning that this is a “polybasic” furin cleavage site), you’d probably look at the rest of the SARS-CoV genomes to estimate a 0.25% chance both arginines being encoded by CGG. If you were asked to guess the maximum fragment length from digestion by common enzymes, you’d estimate less than 0.1% chance of seeing a restriction map exactly in the idealized range for reverse genetic systems AND be produced exclusively by silent mutations AND have P<0.01 significance when testing for higher concentration of silent mutations within those restriction sites compared to the rest of the genome.
You’d combine that information to conservatively estimate less than 1 in 40 billion odds of seeing a SARS-CoV with a human-specific FCS appearing in Wuhan looking like an infectious clone. You would guess the emergence would preferentially infect animal traders, cause a widespread outbreak in animal trade networks with multiple disparate spillover events. You would guess reservoirs would be easily discoverable by sampling animal markets in which outbreaks occur.
If, on the other hand, you had DEFUSE in your hand and were asked to forecast what a lab leak from that research would look like, you’d guess that no animals in wet markets would test positive, animal traders would be just as likely to get infected or leave viruses on the table as vegetable traders, and there would be no geographic trail of infections. The virus might show up in UNC, but, if you knew that the WIV was struggling with their HEPA filters, you would guess the event would occur in sub-standard BSL-2 and BSL-3 facilities in Wuhan. You would expect this SARS-COV would emerge after gain-of-function research with enhanced transmissibility in humans, bearing a human-specific FCS and restriction map of an infectious clone. If a lab origin wasn’t immediately known to be the truth, you would expect everyone involved to act with consciousness of guilt. You would expect the Chinese government to obstruct international investigations and refuse to share critical datasets, researchers at the WIV to lie about whether or not anyone got sick, collaborators on this project to either remain silent, or call lab-origin theories lies and assert themselves as the experts on SARS coronaviruses to obstruct investigations into their own research without disclosing the historic conflict of interest.
If you went back in time to 2018 and used state-of-the-art knowledge to forecast either spillover or a DEFUSE-related lab accident, and you saw the entirety of the evidence today, you would realize immediately that SARS-CoV-2 did not spillover from wildlife to people. It emerged as an accident of normal pre-COVID research on bat coronaviruses. It was not likely a weapon, there is no evidence for malice in the emergence. The same evidence that leads me to believe SARS-CoV-2 is a research product also leads me believe it was an accident of that risky pre-COVID research.
Knowing what I know now, if I were to go back in time to 2018 to preempt the SARS-CoV-2 pandemic, I would shut down gain-of-function research on coronaviruses. In fact, I would shut down gain-of-function research on all animal viruses and immediately encourage talks to foster global coordination on biosafety. I would stop at nothing to prevent the insertion of a furin cleavage site in a SARS-CoV at the Wuhan Institute of Virology. I would fix their HEPA filters.
I believe that would preempt the pandemic and save 18 million lives.
It’s not too late to act. The next pandemic hasn’t happened yet, and if we act we can change the course of history. The purpose of this jury is not to convict anyone, but to examine the all the evidence, stare at the hard truth, and write it accurately into history books. The purpose of this jury is to use our knowledge of the truth to prevent future deaths. I’m not one for retribution - that is not my job today, that’s above my pay grade. I’m just a scientist and a believer in restorative justice. We must not let our emotions get the best of us and go to war over a pandemic, using the release of one of the Four Horsemen to release the others. I implore everyone to encourage our representatives to pursue the truth & seek reconciliation in a global, apartisan way.
Every single one of us from Wuhan to Washington is human. Researchers the whole world over were - and still are - conducting risky virological research. Wuhan labs were the unlucky ones whose broken HEPA filters and leaked viruses caused a pandemic, but the next accident could be in any of our countries. We need to take responsibility to manage the global risks of scientific research. This doesn’t mean we stop funding science, it means we establish guardrails to ensure the risks science takes are worth the reward, and the public has the final say in what risks are and are not worth taking.
There’s no glory in winning this case and demonstrating SARS-CoV-2 emerged from a lab. There’s only sadness. We’ve lived through an historic crisis in which 18 million people died, 60 million faced acute hunger, 100 million kids went into poverty, all because of an accident. As incomprehensibly large as this crisis has been, we’ve witnessed a ripple compared to the wave of power of modern biotechnology.
We now know the urgency of global cooperation on biosafety.
I rest my case.
The way Peter Daszak writes in an e-mail how he wants to feign independence remains amazing to read. Thanks for your excellent long read; very readable for laymen!
Nice to see Australia mentioned.
https://geoffpain.substack.com/p/first-detected-covid19-case-arrived